TCGA PBTA comparison exploration
C. Savonen for ALSF CCDL
2020
Purpose:
Why are TCGA and PBTA data so different and why is TCGA TMB not higher than PBTA as we expected from literature and the results from Grobner et al. 2019
Related issues: Original Issue 3 Issue 257 Draft PR 521
Main three exploratory questions asked in this notebook:
Conclusions from this notebook:
Although it may be partially a caller-specific thing, we also suspect a problem with Lancet’s TCGA calls which is likely because it is all WXS data. This issue is further investigated in the Lancet WXS-WGS analysis notebook and the Lancet padded-unpadded analysis notebook.
Post notes:
It was later determined that the BED files used for these TMB calculations were incorrect See Related Issues:
- 568
- 565
- 564
Usage
To run this from the command line, use:
Rscript -e "rmarkdown::render('analyses/snv-callers/lancet-wxs-tests/explore-tcga-pbta.Rmd',
clean = TRUE)"This assumes you are in the top directory of the repository.
Setup
# Magrittr pipe
`%>%` <- dplyr::`%>%`
# We will need the calculate_tmb function from this script
source(file.path("..", "..", "snv-callers", "util", "tmb_functions.R"))Declare directory paths.
scratch_dir <- file.path("..", "..", "..", "scratch")
data_dir <- file.path("..", "..", "..", "data")
ref_dir <- file.path("..", "ref_files")
plots_dir <- file.path("plots", "tcga-vs-pbta-plots")
if (!dir.exists(plots_dir)) {
dir.create(plots_dir, recursive = TRUE)
}Special functions
Function for setting up PBTA data from the database. These databases were originally created by the run consensus bash script for pbta and run consensus bash script for tcga
set_up_data <- function(data_name,
database_path,
metadata,
is_tcga,
cols_to_keep) {
# Given the name of dataset in the database set up the data with the metadata
#
# Args:
# data_name: A string that is the name of the table in the SQLite file you'd like to pull out.
# database_path: file path to an SQlite file made previously by a run_consensus script
# metadata: associated metadata with this database
# is_tcga: TRUE or FALSE for whether or not this is TCGA data. FALSE = PBTA data
# cols_to_keep: A vector of strings indicating the column names to keep
# Start up connection
con <- DBI::dbConnect(
RSQLite::SQLite(),
database_path
)
# Connect to SQL
df <- dplyr::tbl(con, data_name) %>%
# Only keep the columns we want
dplyr::select(cols_to_keep) %>%
# Turn into data.frame
as.data.frame()
# This step is only needed for TCGA data
if (is_tcga) {
df <- df %>%
# Shorten the Tumor_Sample_Barcode so it matches
dplyr::mutate(Tumor_Sample_Barcode = substr(Tumor_Sample_Barcode, 0, 12))
}
# Tack on the metadata columns we want
df <- df %>%
dplyr::inner_join(
metadata %>%
dplyr::select(
Tumor_Sample_Barcode,
experimental_strategy,
short_histology
)
)
# Disconnect this database
DBI::dbDisconnect(con)
# Return this data.frame
return(df)
}Function for making a combined CDF plot for TMB. This plotting function was adapted from the breaks_cdf_plot function in the chromosomal-instability
tmb_cdf_plot <- function(tmb_df, plot_title) {
# Given a data.frame of TMB data, plot it as a CDF plot and save it as a png.
#
# Args:
# tmb_df: a chromosomal breaks density file path where each sample is
# a row with `samples` and `breaks_count` columns.
# plot_title: to be used for the ggplot2::ggtitle and what it will be saved
# as a png as.
cdf_plot <- tmb_df %>%
as.data.frame() %>%
dplyr::mutate(short_histology = tools::toTitleCase(short_histology)) %>%
# Only plot histologies groups with more than `min_samples` number of samples
dplyr::group_by(short_histology, add = TRUE) %>%
# Only keep groups with this amount of samples
dplyr::filter(dplyr::n() > 5) %>%
# Calculate histology group mean
dplyr::mutate(
hist_mean = mean(tmb),
hist_rank = rank(tmb, ties.method = "first") / dplyr::n(),
sample_size = paste0("n = ", dplyr::n())
) %>%
dplyr::ungroup() %>%
dplyr::mutate(short_histology = reorder(short_histology, hist_mean)) %>%
# Now we will plot these as cummulative distribution plots
ggplot2::ggplot(ggplot2::aes(
x = hist_rank,
y = tmb,
color = datasets
)) +
ggplot2::geom_point() +
# Add summary line for mean
ggplot2::geom_segment(
x = 0, xend = 1, color = "grey",
ggplot2::aes(y = hist_mean, yend = hist_mean)
) +
# Separate by histology
ggplot2::facet_wrap(~ short_histology + sample_size, nrow = 1, strip.position = "bottom") +
ggplot2::theme_classic() +
ggplot2::xlab("") +
ggplot2::ylab("TMB") +
# Transform to log10 make non-log y-axis labels
ggplot2::scale_y_continuous(trans = "log1p", breaks = c(0, 1, 3, 10, 30)) +
ggplot2::scale_x_continuous(limits = c(-0.2, 1.2), breaks = c()) +
# Making it pretty
ggplot2::theme(legend.position = "none") +
ggplot2::theme(
axis.text.x = ggplot2::element_blank(),
axis.ticks.x = ggplot2::element_blank(),
strip.placement = "outside",
strip.text = ggplot2::element_text(size = 10, angle = 90, hjust = 1),
strip.background = ggplot2::element_rect(fill = NA, color = NA)
) +
ggplot2::ggtitle(plot_title)
# Save as a PNG
ggplot2::ggsave(filename = file.path(plots_dir, paste0("tmb-cdf-", plot_title, ".png")),
plot = cdf_plot,
width = 10,
height = 7.5)
}Function for comparing the sequence overlap of two GenomicRanges objects.
bed_overlap <- function(bed_granges_1,
bed_granges_2,
name_1,
name_2,
plot_name) {
# Given two GenomicRanges objects make a VennDiagram of their overlap
# Find intersection
overlaps <- GenomicRanges::intersect(bed_granges_1, bed_granges_2)
# Reduce these ranges for good measure in case they have overlaps
overlaps <- GenomicRanges::reduce(overlaps)
# Percent of TCGA Target region covered by overlap
sum(overlaps@ranges@width) / sum(bed_granges_2@ranges@width)
# Percent of PBTA Target region covered by overlap
sum(bed_granges_1@ranges@width)
# Make filename to save plot as
plot.file <- file.path(plots_dir, paste0(plot_name, ".png"))
# Make the Venn diagram
grid::grid.newpage()
venn.plot <- VennDiagram::draw.pairwise.venn(
area1 = sum(bed_granges_1@ranges@width),
area2 = sum(bed_granges_2@ranges@width),
cross.area = sum(overlaps@ranges@width),
category = c(name_1, name_2),
fill = c("#F8766D", "#00BFC4"),
cex = 2,
cat.cex = 1.5,
ext.pos = 0,
ext.dist = -0.01,
ext.length = .8,
ext.line.lwd = 2,
ext.line.lty = "dashed",
margin = 0.1
)
grid::grid.draw(venn.plot)
# Save as a PNG
png(plot.file)
grid::grid.draw(venn.plot)
dev.off()
# Print out a summary of the ratios
cat(
" Ratio of", name_1, "overlapped:",
sum(overlaps@ranges@width) / sum(bed_granges_1@ranges@width), "\n",
"Ratio of", name_2, "overlapped:",
sum(overlaps@ranges@width) / sum(bed_granges_2@ranges@width), "\n"
)
}Read in the metadata
Do some minor formatting so it works with the set_up_data function.
pbta_metadata <- readr::read_tsv(file.path(data_dir, "pbta-histologies.tsv")) %>%
# MAF files generally call this Tumor_Sample_Barcode and we will need to join by this ID
dplyr::rename(Tumor_Sample_Barcode = Kids_First_Biospecimen_ID)Parsed with column specification:
cols(
.default = col_character(),
OS_days = [32mcol_double()[39m,
age_last_update_days = [32mcol_double()[39m,
normal_fraction = [32mcol_double()[39m,
tumor_fraction = [32mcol_double()[39m,
tumor_ploidy = [32mcol_double()[39m,
molecular_subtype = [33mcol_logical()[39m
)
See spec(...) for full column specifications.
493 parsing failures.
row col expected actual file
2334 molecular_subtype 1/0/T/F/TRUE/FALSE Group3 '../../../data/pbta-histologies.tsv'
2335 molecular_subtype 1/0/T/F/TRUE/FALSE Group4 '../../../data/pbta-histologies.tsv'
2336 molecular_subtype 1/0/T/F/TRUE/FALSE Group3 '../../../data/pbta-histologies.tsv'
2337 molecular_subtype 1/0/T/F/TRUE/FALSE Group3 '../../../data/pbta-histologies.tsv'
2338 molecular_subtype 1/0/T/F/TRUE/FALSE Group3 '../../../data/pbta-histologies.tsv'
.... ................. .................. ...... ....................................
See problems(...) for more details.tcga_metadata <- readr::read_tsv(file.path(data_dir, "pbta-tcga-manifest.tsv")) %>%
dplyr::mutate(
experimental_strategy = "WXS", # This field doesn't exist for this data, but all is WXS
short_histology = Primary_diagnosis
) # This field is named differently in PBTAParsed with column specification:
cols(
Normal_BAM = [31mcol_character()[39m,
Tumor_BAM = [31mcol_character()[39m,
Tumor_Sample_Barcode = [31mcol_character()[39m,
broad_histology = [31mcol_character()[39m,
Primary_diagnosis = [31mcol_character()[39m
)Set up the datasets from the databases
Declare the columns we’ll keep.
cols_to_keep <- c(
"Chromosome",
"Start_Position",
"End_Position",
"Reference_Allele",
"Allele",
"Tumor_Sample_Barcode",
"Variant_Classification",
"t_depth",
"t_ref_count",
"t_alt_count",
"n_depth",
"n_ref_count",
"n_alt_count",
"VAF"
)Set up TCGA data.
tcga_db_file <- file.path(scratch_dir, "tcga_snv_db.sqlite")
tcga_lancet <- set_up_data(data_name = "lancet",
database_path = tcga_db_file,
metadata = tcga_metadata,
is_tcga = TRUE,
cols_to_keep)Joining, by = "Tumor_Sample_Barcode"tcga_mutect <- set_up_data(data_name = "mutect",
database_path = tcga_db_file,
metadata = tcga_metadata,
is_tcga = TRUE,
cols_to_keep)Warning: call dbDisconnect() when finished working with a connection
Joining, by = "Tumor_Sample_Barcode"tcga_strelka <- set_up_data(data_name ="strelka",
database_path = tcga_db_file,
metadata = tcga_metadata,
is_tcga = TRUE,
cols_to_keep)Joining, by = "Tumor_Sample_Barcode"tcga_consensus <- set_up_data("consensus",
database_path = tcga_db_file,
metadata = tcga_metadata,
is_tcga = TRUE,
cols_to_keep)Joining, by = "Tumor_Sample_Barcode"Set up PBTA data.
pbta_db_file <- file.path(scratch_dir, "snv_db.sqlite")
pbta_lancet <- set_up_data(data_name = "lancet",
database_path = pbta_db_file,
metadata = pbta_metadata,
is_tcga = FALSE,
cols_to_keep)How does the tumor sequencing depth compare for each TCGA and PBTA data?
Set up combined PBTA and TCGA data.frames.
lancet <- dplyr::bind_rows(list(
tcga = tcga_lancet,
pbta = pbta_lancet
), .id = "dataset") %>%
dplyr::mutate(dataset = as.factor(dataset))
mutect <- dplyr::bind_rows(list(
tcga = tcga_mutect,
pbta = pbta_mutect
), .id = "dataset") %>%
dplyr::mutate(dataset = as.factor(dataset))
strelka <- dplyr::bind_rows(list(
tcga = tcga_strelka,
pbta = pbta_strelka
), .id = "dataset") %>%
dplyr::mutate(dataset = as.factor(dataset))We’ll plot the tumor sequencing depth for each caller as a series of density plots.
lancet %>%
ggplot2::ggplot(ggplot2::aes(x = log10(t_depth), y = ..scaled..,
color = dataset)) +
ggplot2::geom_density(bw = 0.5)
mutect %>%
ggplot2::ggplot(ggplot2::aes(x = log10(t_depth), y = ..scaled..,
color = dataset)) +
ggplot2::geom_density(bw = 0.2)
strelka %>%
ggplot2::ggplot(ggplot2::aes(x = log10(t_depth), y = ..scaled.., color = dataset)) +
ggplot2::geom_density(bw = 0.2)
How does the TMB comparison look for each caller by itself?
Lancet only TMB:
lancet_tmb <- dplyr::bind_rows(list(
pbta = calculate_tmb(pbta_lancet,
bed_wgs = file.path(data_dir, "intersect_cds_lancet_strelka_mutect_WGS.bed"),
bed_wxs = file.path(data_dir, "intersect_cds_lancet_WXS.bed")
),
tcga = calculate_tmb(tcga_lancet,
bed_wgs = file.path(data_dir, "intersect_cds_lancet_strelka_mutect_WGS.bed"), # There's no TCGA WGS samples, this is just a place holder and won't be used.
bed_wxs = file.path(ref_dir, "intersect_cds_gencode_liftover_WXS.bed")
)
), .id = "datasets")Parsed with column specification:
cols(
X1 = [31mcol_character()[39m,
X2 = [32mcol_double()[39m,
X3 = [32mcol_double()[39m
)
Parsed with column specification:
cols(
X1 = [31mcol_character()[39m,
X2 = [32mcol_double()[39m,
X3 = [32mcol_double()[39m
)Ratio of variants in this BED: 1.739157
Ratio of variants being filtered out: -0.7391572
Ratio of variants in this BED: 1.651031
Ratio of variants being filtered out: -0.6510313 Unreplaced values treated as NA as .x is not compatible. Please specify replacements exhaustively or supply .defaultParsed with column specification:
cols(
X1 = [31mcol_character()[39m,
X2 = [32mcol_double()[39m,
X3 = [32mcol_double()[39m
)
Parsed with column specification:
cols(
X1 = [31mcol_character()[39m,
X2 = [32mcol_double()[39m,
X3 = [32mcol_double()[39m
)Ratio of variants in this BED: 0.7421131
Ratio of variants being filtered out: 0.2578869
Ratio of variants in this BED: NaN
Ratio of variants being filtered out: NaN tmb_cdf_plot(lancet_tmb, plot_title = "Lancet")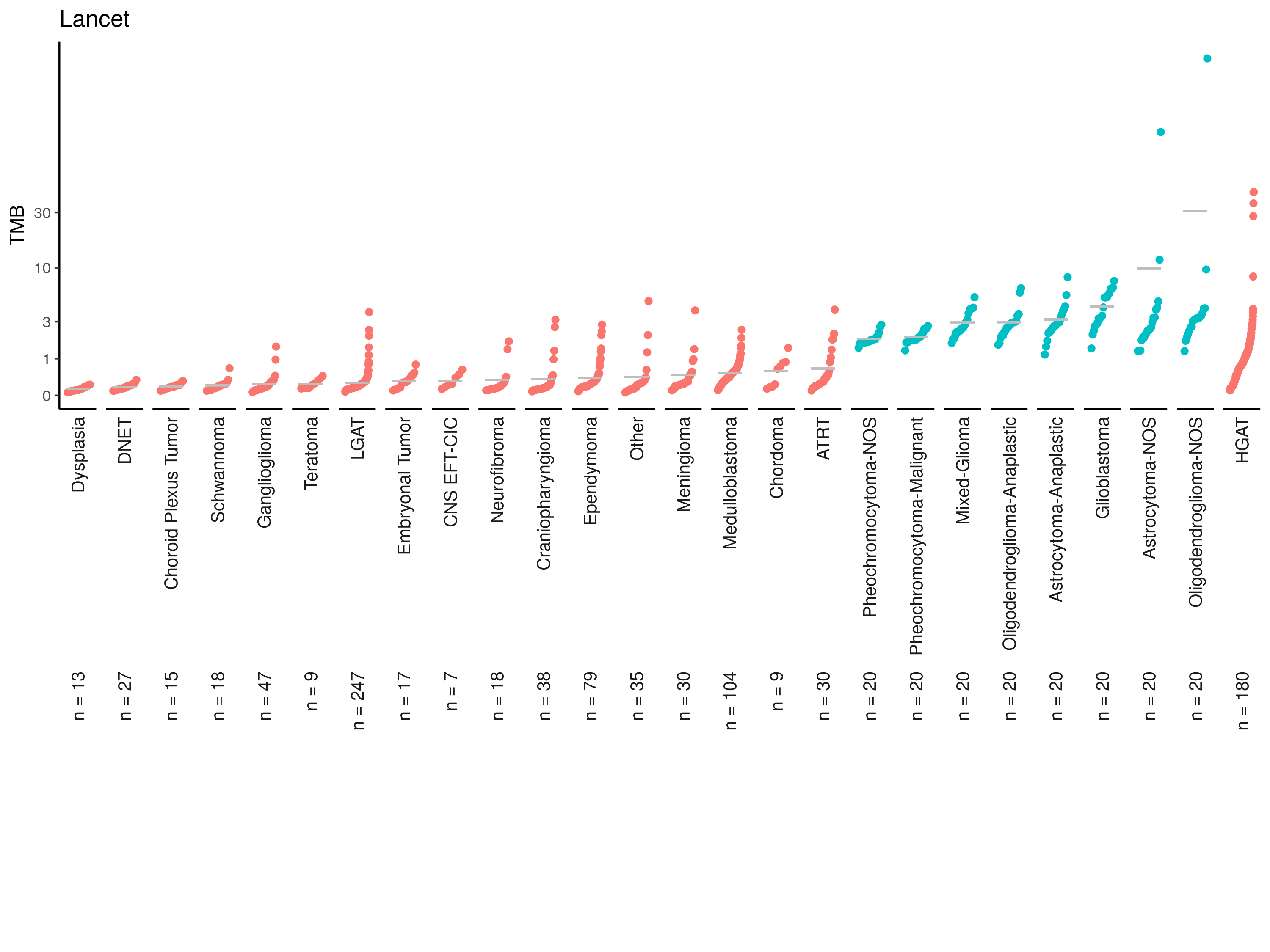{kind=link}
Mutect only TMB:
mutect_tmb <- dplyr::bind_rows(list(
pbta = calculate_tmb(pbta_mutect,
bed_wgs = file.path(data_dir, "intersect_cds_lancet_strelka_mutect_WGS.bed"),
bed_wxs = file.path(data_dir, "intersect_cds_lancet_WXS.bed")
),
tcga = calculate_tmb(tcga_mutect,
bed_wgs = file.path(data_dir, "intersect_cds_lancet_strelka_mutect_WGS.bed"),
bed_wxs = file.path(ref_dir, "intersect_cds_gencode_liftover_WXS.bed")
)
), .id = "datasets")Parsed with column specification:
cols(
X1 = [31mcol_character()[39m,
X2 = [32mcol_double()[39m,
X3 = [32mcol_double()[39m
)
Parsed with column specification:
cols(
X1 = [31mcol_character()[39m,
X2 = [32mcol_double()[39m,
X3 = [32mcol_double()[39m
)Ratio of variants in this BED: 1.98804
Ratio of variants being filtered out: -0.9880405
Ratio of variants in this BED: 1.091582
Ratio of variants being filtered out: -0.09158185 Unreplaced values treated as NA as .x is not compatible. Please specify replacements exhaustively or supply .defaultParsed with column specification:
cols(
X1 = [31mcol_character()[39m,
X2 = [32mcol_double()[39m,
X3 = [32mcol_double()[39m
)
Parsed with column specification:
cols(
X1 = [31mcol_character()[39m,
X2 = [32mcol_double()[39m,
X3 = [32mcol_double()[39m
)Ratio of variants in this BED: 0.4940234
Ratio of variants being filtered out: 0.5059766
Ratio of variants in this BED: NaN
Ratio of variants being filtered out: NaN # Make a CDF plot
tmb_cdf_plot(mutect_tmb, plot_title = "Mutect2")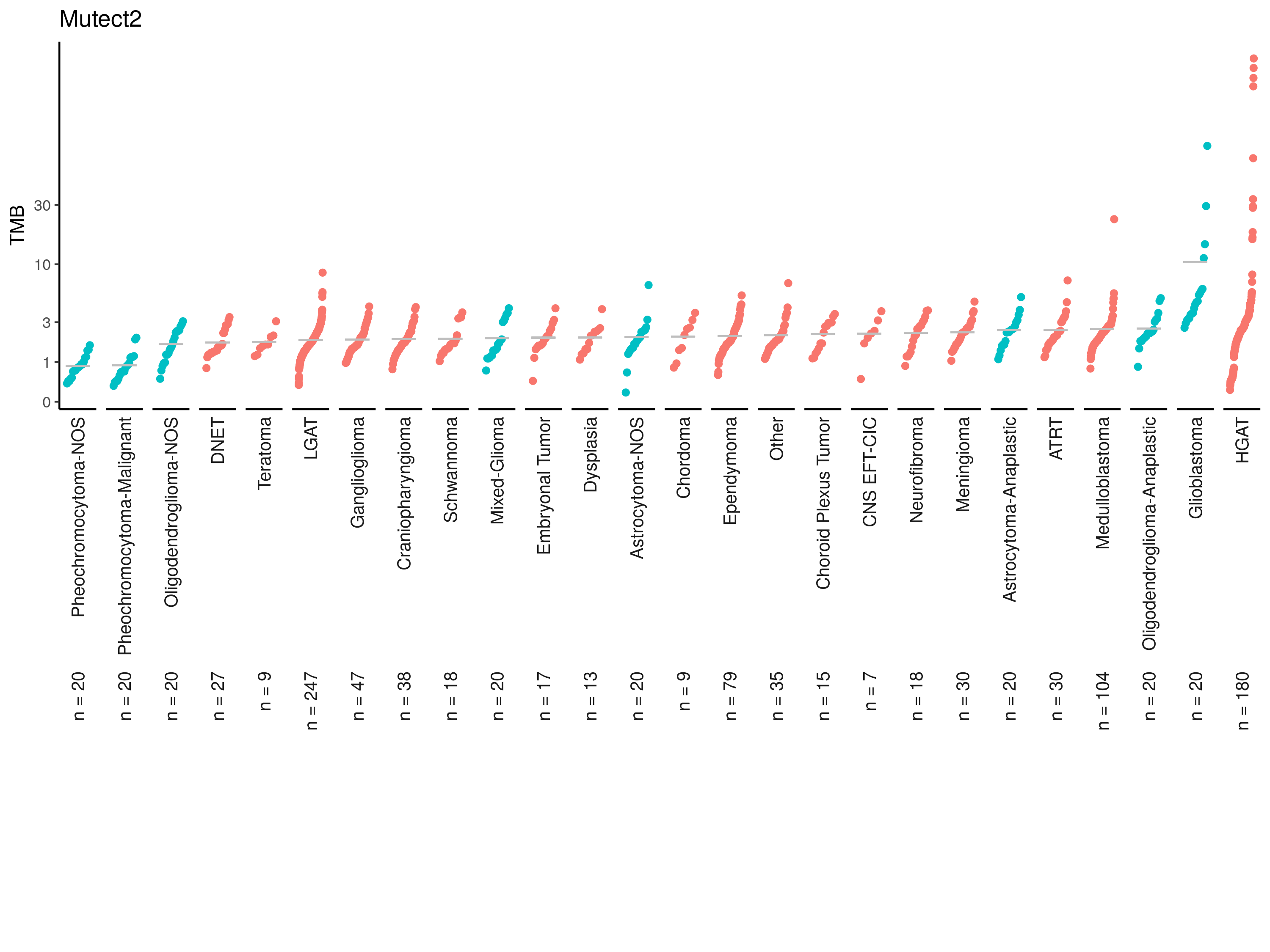{kind=link}
Strelka only TMB:
strelka_tmb <- dplyr::bind_rows(list(
pbta = calculate_tmb(pbta_strelka,
bed_wgs = file.path(data_dir, "intersect_cds_lancet_strelka_mutect_WGS.bed"),
bed_wxs = file.path(data_dir, "intersect_cds_lancet_WXS.bed")
),
tcga = calculate_tmb(tcga_strelka,
bed_wgs = file.path(data_dir, "intersect_cds_lancet_strelka_mutect_WGS.bed"),
bed_wxs = file.path(ref_dir, "intersect_cds_gencode_liftover_WXS.bed")
)
), .id = "datasets")Parsed with column specification:
cols(
X1 = [31mcol_character()[39m,
X2 = [32mcol_double()[39m,
X3 = [32mcol_double()[39m
)
Parsed with column specification:
cols(
X1 = [31mcol_character()[39m,
X2 = [32mcol_double()[39m,
X3 = [32mcol_double()[39m
)Ratio of variants in this BED: 1.701877
Ratio of variants being filtered out: -0.7018769
Ratio of variants in this BED: 1.110021
Ratio of variants being filtered out: -0.1100208 Unreplaced values treated as NA as .x is not compatible. Please specify replacements exhaustively or supply .defaultParsed with column specification:
cols(
X1 = [31mcol_character()[39m,
X2 = [32mcol_double()[39m,
X3 = [32mcol_double()[39m
)
Parsed with column specification:
cols(
X1 = [31mcol_character()[39m,
X2 = [32mcol_double()[39m,
X3 = [32mcol_double()[39m
)Ratio of variants in this BED: 0.4836536
Ratio of variants being filtered out: 0.5163464
Ratio of variants in this BED: NaN
Ratio of variants being filtered out: NaN # Make a CDF plot
tmb_cdf_plot(strelka_tmb, plot_title = "Strelka2")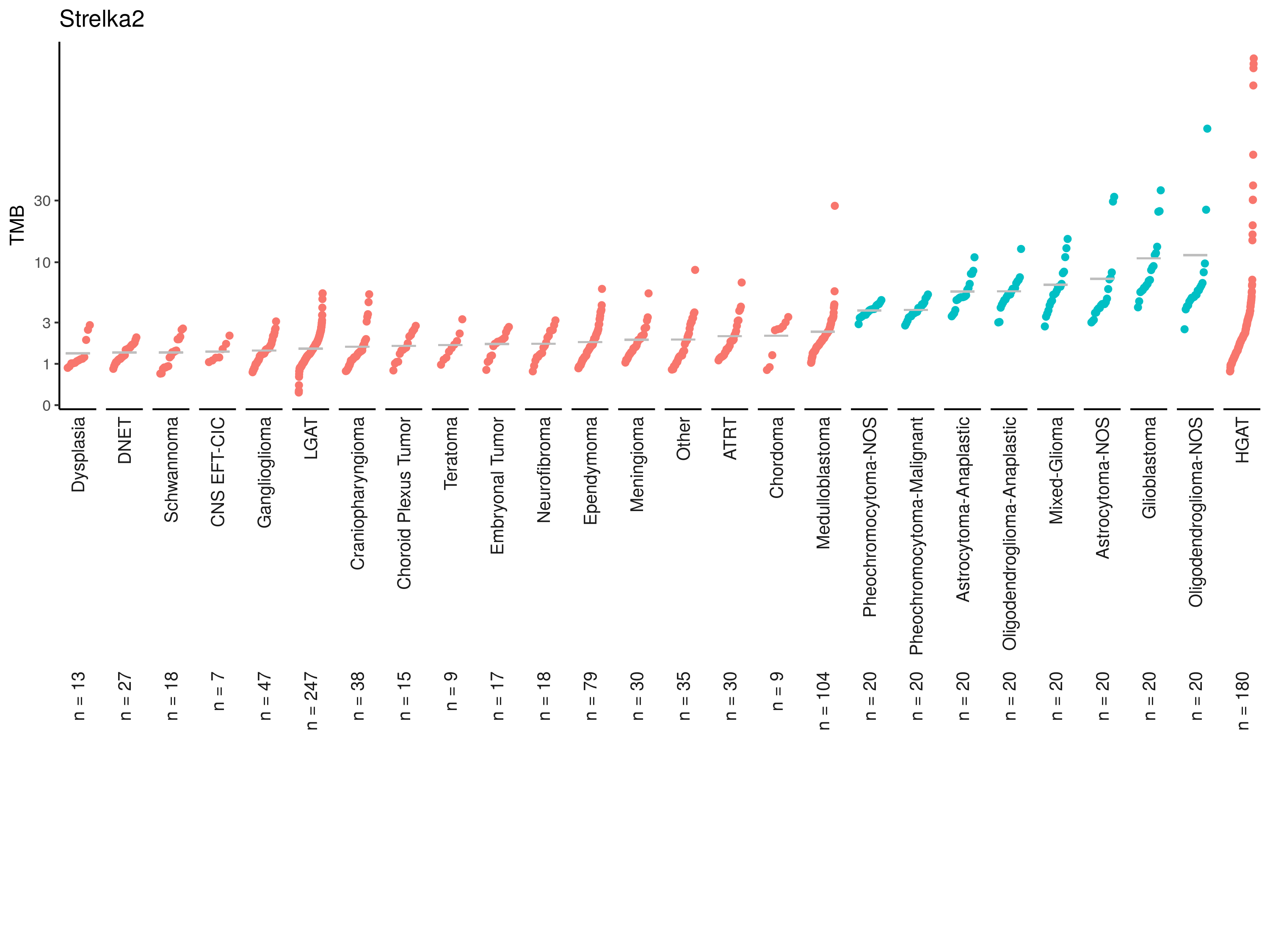{kind=link}
Consensus TMB:
consensus_tmb <- dplyr::bind_rows(list(
pbta = calculate_tmb(pbta_consensus,
bed_wgs = file.path(data_dir, "intersect_cds_lancet_strelka_mutect_WGS.bed"),
bed_wxs = file.path(data_dir, "intersect_cds_lancet_WXS.bed")
),
tcga = calculate_tmb(tcga_consensus,
bed_wgs = file.path(data_dir, "intersect_cds_lancet_strelka_mutect_WGS.bed"),
bed_wxs = file.path(ref_dir, "intersect_cds_gencode_liftover_WXS.bed")
)
), .id = "datasets")Parsed with column specification:
cols(
X1 = [31mcol_character()[39m,
X2 = [32mcol_double()[39m,
X3 = [32mcol_double()[39m
)
Parsed with column specification:
cols(
X1 = [31mcol_character()[39m,
X2 = [32mcol_double()[39m,
X3 = [32mcol_double()[39m
)Ratio of variants in this BED: 3.093321
Ratio of variants being filtered out: -2.093321
Ratio of variants in this BED: 1.427508
Ratio of variants being filtered out: -0.4275085 Unreplaced values treated as NA as .x is not compatible. Please specify replacements exhaustively or supply .defaultParsed with column specification:
cols(
X1 = [31mcol_character()[39m,
X2 = [32mcol_double()[39m,
X3 = [32mcol_double()[39m
)
Parsed with column specification:
cols(
X1 = [31mcol_character()[39m,
X2 = [32mcol_double()[39m,
X3 = [32mcol_double()[39m
)Ratio of variants in this BED: 0.7455764
Ratio of variants being filtered out: 0.2544236
Ratio of variants in this BED: NaN
Ratio of variants being filtered out: NaN # Make a CDF plot
tmb_cdf_plot(consensus_tmb, plot_title = "Consensus")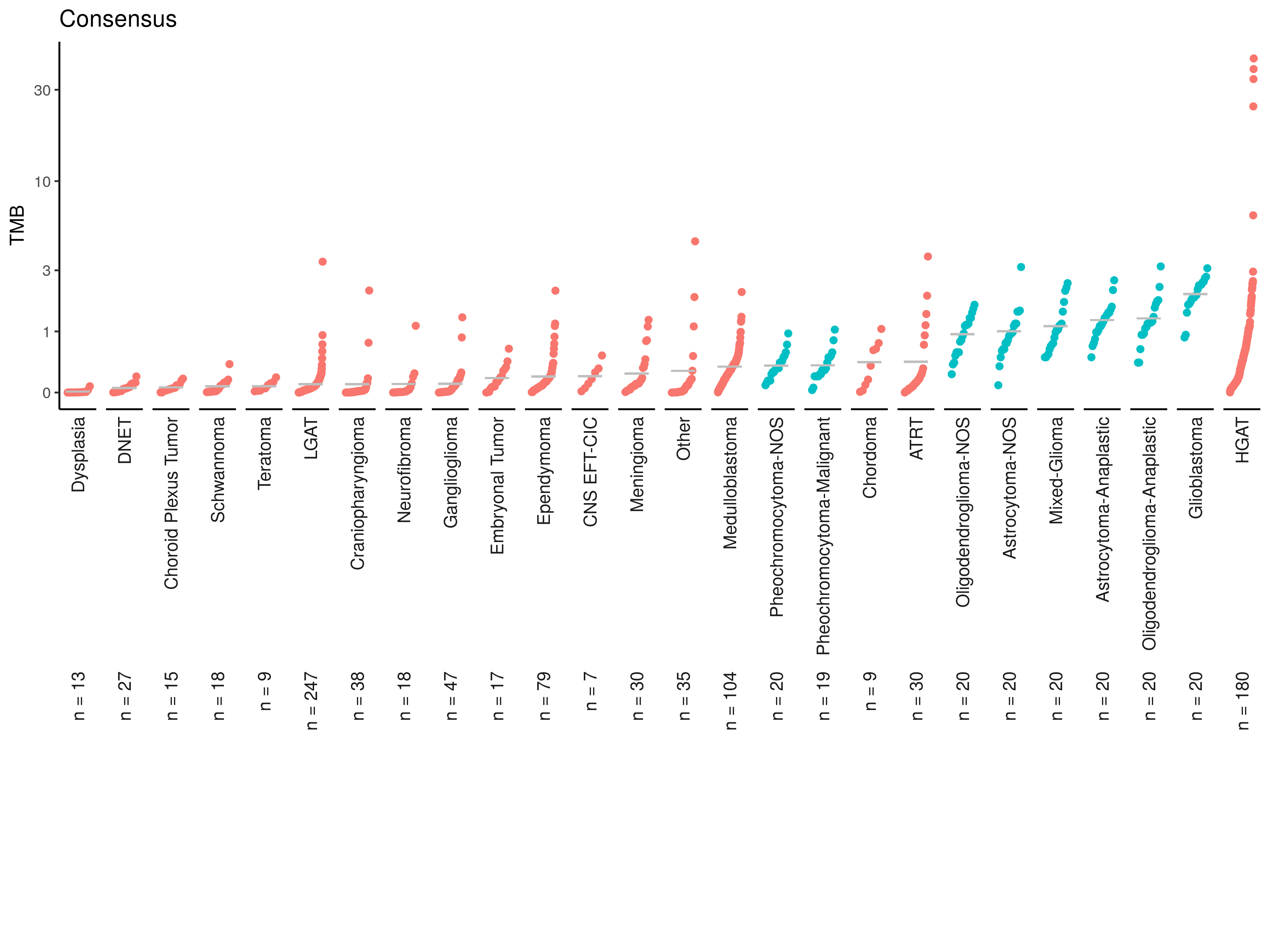{kind=link}
Overlap of the target regions of both datasets
Download a TCGA Target BED regions file from MC3, and format the chromosome data to be chr, save as a TSV file.
tcga_bed <- readr::read_tsv("https://api.gdc.cancer.gov/data/7f0d3ab9-8bef-4e3b-928a-6090caae885b", col_names = c("chr", "start", "end")) %>%
dplyr::filter(!is.na(chr)) %>%
dplyr::mutate(chr = paste0("chr", chr)) %>%
readr::write_tsv(file.path(ref_dir, "gencode.v19.basic.exome.tsv"),
col_names = FALSE
)Parsed with column specification:
cols(
chr = [32mcol_double()[39m,
start = [32mcol_double()[39m,
end = [32mcol_double()[39m
)
27285 parsing failures.
row col expected actual file
697048 chr a double MT 'https://api.gdc.cancer.gov/data/7f0d3ab9-8bef-4e3b-928a-6090caae885b'
697049 chr a double MT 'https://api.gdc.cancer.gov/data/7f0d3ab9-8bef-4e3b-928a-6090caae885b'
697050 chr a double MT 'https://api.gdc.cancer.gov/data/7f0d3ab9-8bef-4e3b-928a-6090caae885b'
697051 chr a double MT 'https://api.gdc.cancer.gov/data/7f0d3ab9-8bef-4e3b-928a-6090caae885b'
697052 chr a double MT 'https://api.gdc.cancer.gov/data/7f0d3ab9-8bef-4e3b-928a-6090caae885b'
...... ... ........ ...... ......................................................................
See problems(...) for more details.After using UCSC BED liftover to convert from the Target BED regions for TCGA data from hg19 to hg38 (What the PBTA data is using).
tcga_lift_bed <- readr::read_tsv(file.path(
ref_dir,
"hg38_liftover_genome_gencode.v19.basic.exome.bed"
),
col_names = c("chr", "start", "end")
) %>%
dplyr::mutate(chr = stringr::word(chr, sep = "_", 1))Parsed with column specification:
cols(
chr = [31mcol_character()[39m,
start = [32mcol_double()[39m,
end = [32mcol_double()[39m
)# Make GRanges for CNV data
tcga_lift_granges <- GenomicRanges::GRanges(
seqnames = tcga_lift_bed$chr,
ranges = IRanges::IRanges(
start = tcga_lift_bed$start,
end = tcga_lift_bed$end
)
)
# Reduce this to it's essential ranges
tcga_lift_granges <- GenomicRanges::reduce(tcga_lift_granges)Make a TCGA BED GenomicRanges.
# Make GRanges for CNV data
tcga_granges <- GenomicRanges::GRanges(
seqnames = tcga_bed$chr,
ranges = IRanges::IRanges(
start = tcga_bed$start,
end = tcga_bed$end
)
)
# Reduce this to it's essential ranges
tcga_granges <- GenomicRanges::reduce(tcga_granges)Format the PBTA WXS data as a GRanges object.
# pbta_bed <- readr::read_tsv(file.path(scratch_dir, "intersect_cds_WXS.bed"),
pbta_bed <- readr::read_tsv(file.path(data_dir, "WXS.hg38.100bp_padded.bed"),
col_names = c("chr", "start", "end")
) %>%
dplyr::filter(!is.na(chr))Parsed with column specification:
cols(
chr = [31mcol_character()[39m,
start = [32mcol_double()[39m,
end = [32mcol_double()[39m
)# Make GRanges for CNV data
pbta_granges <- GenomicRanges::GRanges(
seqnames = pbta_bed$chr,
ranges = IRanges::IRanges(
start = pbta_bed$start,
end = pbta_bed$end
)
)
# Reduce this to it's essential ranges
pbta_granges <- GenomicRanges::reduce(pbta_granges)Read in the coding sequence regions file.
cds_bed <- readr::read_tsv(file.path(
scratch_dir,
"gencode.v27.primary_assembly.annotation.bed"
),
col_names = c("chr", "start", "end")
) %>%
dplyr::filter(!is.na(chr))Parsed with column specification:
cols(
chr = [31mcol_character()[39m,
start = [32mcol_double()[39m,
end = [32mcol_double()[39m
)# Make GRanges
cds_granges <- GenomicRanges::GRanges(
seqnames = cds_bed$chr,
ranges = IRanges::IRanges(
start = cds_bed$start,
end = cds_bed$end
)
)
# Reduce this to it's essential ranges
cds_granges <- GenomicRanges::reduce(cds_granges)We also need to see how this translates to CDS regions:
# Find CDS intersection
pbta_cds_granges <- GenomicRanges::intersect(pbta_granges, cds_granges)Each of the 2 combined objects has sequence levels not in the other:
- in 'x': chr7_KI270803v1_alt, chr15_KI270850v1_alt, chr17_KI270909v1_alt, chr19_KI270938v1_alt, chr22_KI270879v1_alt, chrUn_GL000213v1
- in 'y': chrM, GL000009.2, GL000194.1, GL000195.1, GL000205.2, GL000213.1, GL000218.1, GL000219.1, KI270711.1, KI270713.1, KI270721.1, KI270726.1, KI270727.1, KI270728.1, KI270731.1, KI270734.1
Make sure to always combine/compare objects based on the same reference
genome (use suppressWarnings() to suppress this warning).tcga_cds_granges <- GenomicRanges::intersect(tcga_granges, cds_granges)
tcga_lift_cds_granges <- GenomicRanges::intersect(tcga_lift_granges, cds_granges)Each of the 2 combined objects has sequence levels not in the other:
- in 'x': chrUn
- in 'y': chrM, chrX, chrY, GL000009.2, GL000194.1, GL000195.1, GL000205.2, GL000213.1, GL000218.1, GL000219.1, KI270711.1, KI270713.1, KI270721.1, KI270726.1, KI270727.1, KI270728.1, KI270731.1, KI270734.1
Make sure to always combine/compare objects based on the same reference
genome (use suppressWarnings() to suppress this warning).Make Venn diagrams of these overlaps
Find overlap between the TCGA (non-liftover) and PBTA data.
bed_overlap(pbta_granges,
tcga_granges,
plot_name = "PBTA vs TCGA WXS target BED",
name_1 = "PBTA",
name_2 = "TCGA"
) Ratio of PBTA overlapped: 0.07363001
Ratio of TCGA overlapped: 0.1186468 
Find overlap between the liftover TCGA target BED region and PBTA data.
bed_overlap(pbta_granges,
tcga_lift_granges,
plot_name = "PBTA vs TCGA WXS liftover target BED",
name_1 = "PBTA",
name_2 = "TCGA liftover"
)Each of the 2 combined objects has sequence levels not in the other:
- in 'x': chr7_KI270803v1_alt, chr15_KI270850v1_alt, chr17_KI270909v1_alt, chr19_KI270938v1_alt, chr22_KI270879v1_alt, chrUn_GL000213v1, chrX, chrY
- in 'y': chrUn
Make sure to always combine/compare objects based on the same reference
genome (use suppressWarnings() to suppress this warning). Ratio of PBTA overlapped: 0.487083
Ratio of TCGA liftover overlapped: 0.7862399 
Find overlap between coding sequences only of the liftover TCGA target BED region and PBTA data.
bed_overlap(pbta_cds_granges,
tcga_lift_cds_granges,
plot_name = "PBTA vs TCGA WXS liftover coding sequence target BED",
name_1 = "PBTA CDS",
name_2 = "TCGA lift CDS"
)Each of the 2 combined objects has sequence levels not in the other:
- in 'x': chr7_KI270803v1_alt, chr15_KI270850v1_alt, chr17_KI270909v1_alt, chr19_KI270938v1_alt, chr22_KI270879v1_alt, chrUn_GL000213v1
- in 'y': chrUn
Make sure to always combine/compare objects based on the same reference
genome (use suppressWarnings() to suppress this warning). Ratio of PBTA CDS overlapped: 0.942882
Ratio of TCGA lift CDS overlapped: 0.9794732 
Find overlap between coding sequences only of the TCGA target BED region and PBTA data.
bed_overlap(pbta_cds_granges,
tcga_cds_granges,
plot_name = "PBTA vs TCGA WXS Coding sequence target BED",
name_1 = "PBTA CDS",
name_2 = "TCGA CDS"
) Ratio of PBTA CDS overlapped: 0.08945405
Ratio of TCGA CDS overlapped: 0.975733 
Session Info
sessionInfo()R version 3.6.0 (2019-04-26)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Debian GNU/Linux 9 (stretch)
Matrix products: default
BLAS/LAPACK: /usr/lib/libopenblasp-r0.2.19.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C LC_TIME=en_US.UTF-8
[4] LC_COLLATE=en_US.UTF-8 LC_MONETARY=en_US.UTF-8 LC_MESSAGES=C
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C LC_ADDRESS=C
[10] LC_TELEPHONE=C LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets methods base
other attached packages:
[1] GenomicRanges_1.38.0 GenomeInfoDb_1.22.0 IRanges_2.20.1 S4Vectors_0.24.1
[5] BiocGenerics_0.32.0
loaded via a namespace (and not attached):
[1] tidyselect_0.2.5 xfun_0.8 purrr_0.3.2 colorspace_1.4-1
[5] htmltools_0.3.6 base64enc_0.1-3 yaml_2.2.0 blob_1.1.1
[9] rlang_0.4.0 pillar_1.4.2 glue_1.3.1 DBI_1.0.0
[13] bit64_0.9-7 dbplyr_1.4.2 lambda.r_1.2.3 GenomeInfoDbData_1.2.2
[17] stringr_1.4.0 zlibbioc_1.32.0 munsell_0.5.0 gtable_0.3.0
[21] futile.logger_1.4.3 memoise_1.1.0 evaluate_0.14 labeling_0.3
[25] knitr_1.23 curl_3.3 Rcpp_1.0.1 readr_1.3.1
[29] scales_1.0.0 formatR_1.7 jsonlite_1.6 XVector_0.26.0
[33] bit_1.1-14 ggplot2_3.2.0 hms_0.4.2 digest_0.6.20
[37] stringi_1.4.3 dplyr_0.8.3 grid_3.6.0 tools_3.6.0
[41] bitops_1.0-6 magrittr_1.5 RCurl_1.95-4.12 lazyeval_0.2.2
[45] tibble_2.1.3 RSQLite_2.1.1 futile.options_1.0.1 crayon_1.3.4
[49] pkgconfig_2.0.2 data.table_1.12.2 assertthat_0.2.1 rmarkdown_1.13
[53] rstudioapi_0.10 R6_2.4.0 VennDiagram_1.6.20 compiler_3.6.0